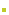第一章 总 则
第一条根据《中华人民共和国药品管理法V以下称《药品管理法口的规定,制定本办法。
第二条本办法适用于所有有关药品生产、经营、使用、检验、科研的单位和个人。
军队的药品生产企业生产民用药品的,适用本办法。
第三条药品的生产、经营,应当把社会效益放在第一位,严禁生产、经营、使用假药或者劣药。严禁未经许可生产、经营药品和配制制剂。
第二章 药品监督管理职责
第四条国务院卫生行政部门主管全国药品监督管理工作,其主要职责是:
(一)执行《药品管理法》及本办法;
(二)起草有关药品监督管理的法规、制定配套的单行办法;
(三)颁布《中国药典》和药品标准;
(四)审批新药、核发药品批准文号;
(五)对药品的生产、经营、使用进行监督;
(六)组织对已经生产的药品的药效、副作用进行调查再评价,并及时提供和公布有关质量方面的资料;
(七)依照《药品管理法》和本办法决定行政处罚。
县以上地方各级卫生行政部门的药政机构主管所辖行政区的药品监督管理工作。
第五条县级以上卫生行政部门设置的药品检验所,在同级卫生行政部门的领导下,按照国家药品标准和省、自治区、直辖市的药品标准对药品进行检验。
第六条县级以上卫生行政部门设药品监督员,国家药品监督员由国务院卫生行政部门审核发给证书;省、自治区、直辖市药品监督员和自治州、市或者县的药品监督员由卫生行政部门提名,同级人民政府审核发给证书。
药品监督员的职责由国务院卫生行政部门另行规定。
第七条药品监督员在履行职责时,应出示证件,按照国家有关规定抽取样品和索取有关资料并开具清单。对药品生产企业和科研单位提供的保密的技术资料,应当承提保密责任。
药品监督员对暂行封存待处理的药品,应注明封存期限,该期限一般不得超过15天。
第三章 审核批准许可证的程序
第八条《药品管理法》第四条第一款规定的审批程序,是指开办药品生产企业(包括各种形式的联营、中外合资企业、中外合作企业以及外资企业),除按照国家规定履行其本建设报批程序以外,必须依次履行下列程序:
(一)由企业或者企业的上级部门向所在省、自治区、直辖市的药品生产经营主管部门申报,经审查同意后,送同级卫生行政部门;
(二)经所在省、自治区、直辖市卫生行政部门审核批准,发给《药品生产企业许可证》。
药品生产经营主管部门和卫生行政部门应当在各自收到全部申报材料后的30日内,作出是否同意或者批准的决定。
第九条药品生产企业另设分厂或者在厂区外另设车间的,由药品生产企业向分厂或者车间所在地的省、自治区、直辖市药品生产经营主管部门申请,经审查同意后,送同级卫生行政部门申请办理《药品生产企业许可证》。《药品生产企业许可证》应注明分厂(车间)和生产范围。
第十条《药品管理法》第十条第一款规定的审批程序,是指药品经营企业(包括专营或者兼营的批发、零售商店或者公司)按照以下规定申请办理《药品经营企业许可证》:
(一)经营药品批发业务的企业,由省、自治区、直辖市的药品生产经营主管部门审查同意,经省、自治区、直辖市卫生行政部门审核批准,发给《药品经营企业许可证》;
(二)经营药品零售业务的企业,由所在地的自治州、市或者县的药品生产经营主管部门审查同意,经同级卫生行政部门审核批准,发给《药品经营企业许可证》。
药品生产经营主管部门和卫生行政部门应当在各自收到全部申报材料后的30日内,作出是否同意或者批准的决定。
第十一条《药品管理法》第四条、第十条、第二十二条所称“药品生产经营主管部门”,是指县以上地方各级医药归口管理部门或者人民政府指定的部门。
第十二条医疗单位自配制剂,必须向所在省、自治区、直辖市卫生行政部门申请,经审查批准后发给《制剂许可证》。
受理审查的卫生行政部门应当在收到全部申报材料后30日内,作出是否批准的决定。
第十三条《药品生产企业许可证》、《药品经营企业许可证》、《制剂许可证》的有效期为5年1期满后继续生产)经营药品或者配制制剂的,持证单位应当在期满前6个月重新申请,重新申请的程序与第一次申请的程序相同。
企业破产或者关闭,上述许可证应当由原发证部门缴销。
第十四条《药品生产企业许可证》、《药品经营企业许可证》、《制剂许可证》由国务院卫生行政部门统一印制。
第四章 新药的审批
第十五条国家鼓励研究、创制新药,凡有条件的药品研究单位、高等院校、药品生产企业、医疗单位或者个人都可以从事新药的研究、创制。
第十六条新药审批办法由国务院卫生行政部门制定。
第十七条新药研制单位申请进行新药临床试验,必须按照新药审批办法的规定,报送有关资料和样品。
第十八条新药临床试验或者临床验证,应当在省,自治区、直辖市卫生行政部门批准的医疗单位进行。
第十九条完成临床试验或者临床验证并通过所在省、自治区、直辖市卫生行政部门初审的新药,由研制单位报国务院卫生行政部门审批。经国务院卫生行政部门批准后,发给新药证书。
国务院卫生行政部门应当在收到全部申报材料后,尽快组织药品审评委员会审评,并在审评后的两个月以内,作出是否批准的决定。
第二十条国务院卫生行政部门和省、自治区、直辖市卫生行政部门可以成立药品审评委员会,委员会成员由医疗、科研、生产、教学等方面的医学、药学专家组成。
第二十一条对于新药研制单位或者个人提交的有关资料、数据、工艺等,临床试验或者验证单位、审批部门及其工作人员应当承担保密责任。