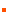һ���ۼ��ع���
1. ������������Ƥ����������Ů������ֳ���Ա��������ԭ������55������һ�ֳ�Ⱦɫ�������Ŵ�������Ⱦɫ��Ϊ46,XX������Ϊ�����ѳ������ӹ������ѹܡ���Ҫ������������Ƥ���ںϳ���̴����ع�����ȱ��ij��ø������������ۼ��أ����г�������21��11�ǻ�øȱ��������ֳ�������в�ͬ�̶ȵ����Ի���Prader�����Ի��̶Ƚ�������Ϊ5�ͣ����߽������Դ����صĿ��з��������������ң��������ԣ�����������غ��[1]����������������������Ƥ����������Ϊͬ�������졣
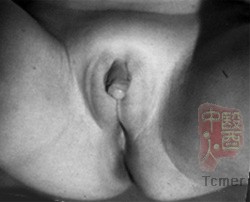
2����������Դ���ۼ��ع�������������Ⱦɫ��Ϊ46,XX����ĸ������Ӥ���ж��������ڷ��ô�����ۼ���[2]������Ϊ������С�������ںϣ������������٣�ͼ1��������ֳ�����Ի��̶���������ҩʱ�䡢���ࡢ����������ʱ���йء���̥������ֳ���ֻ�ǰ��ʹ����ֳ�����Ի����ۼ�������ǿ�����������Ի������ء�
ͼ1����������Դ���ۼ��ع����������������������ںϡ�
1.����ȫ���ۼ��ز������ۺ���: ����28������һ�ֽ�Ϊ���������Ե������Է����쳣������Ϊ46,XY��˫������Ϊغ�裬غͪ���������������ۼ��������쳣�����ۼ��ص�����ЧӦȫ����ɥʧ���ۼ��ع���ȫ��ɥʧ����ΪŮ������ֳ�������ӹ�������ɥʧ����ֳ����������������������Ƥ�������ĸ��ڱ��֣�ͼ2�����ۼ����������λ��XȾɫ��ij��ۣ�Xq11-12�������ѷ����ۼ����������DNA��������ۼ��ؽ�����ĸ���ȱʧ��ͻ���ǵ�����ȫ���ۼ��ز������ۺ�������Ҫԭ��[3]�����ڲ���ȫ���ۼ��ز������ۺ����У�����δ�����ۼ�������������ķ���ȱ�ݣ�������ı���Ҫ�ڷDZ��������������3��δ�������������йص�ת¼�������ӡ�����ȫ���ۼ��ز������ۺ�����ȷ������������Ƥ���ۼ������������IJⶨ[4��5]���ۼ�������������ת¼�������ӵķ���[6]��
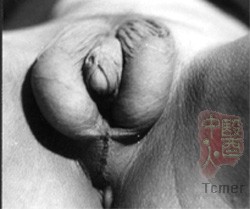
���⣬�������ۼ��غϳ�ȱ�ݶ���ȫȱ��������ֳ��������Ϊ��ȫŮ�ԣ�������ȱ�������Ϊ���Ի����㣬����5��ø�漰�ӵ��̴��ϳ�غͪ����Ժ���ռ�7��17�����ǻ�øȱ�����ߣ�����ֳ��������ΪŮ�ԣ������Ի����֣���Ϊ��ȫ��ȱ��[7]���������ۼ��غϳɲ���ȱ�ݵIJ������������ijЩø��������ļ����йء�
2��غ���˻�����Ϊ�ټ�������3����Ⱦɫ��Ϊ46,XY������ֳ������Ϊ����غͪ��Ӱ�죬�����ںϳ����ң���������������������ٸ�������������̥���ڵı��֣��䲡��Ϊ��̥��غ���˻������ٷ���غͪ������ֳ��δ��һ��������
�������ٷֻ��쳣
�κ��쳣����غ��ֻ��ӳ١���ȫ�Գ��߾��ɵ�������ֳ���Ա��塣
1. �����Ի���: ������13���������������پ����ѳ���غ������������֯�����ٿ����ǵ������ѳ���غ�裬��������ѳ���غ����ͬһ�����ڣ���غ���������Ի������ѳ���غ��һ��ͬʱ�ֻ������й��ܣ�غ��ֻӰ��ͬ����ֳ���ķֻ���������Ϊ��غ�������Ϲܶ����������ơ�����ֳ������̬�仯�ܴ�һ�����Ϊ�������������ԣ���������ѡ����������Ҽ����٣�ͼ3��������̥����غ������ã�������������������������������������������鷿���������ֿ����¾�������Ѫ������̥��غ�����ò��㣬����ʱ���Һ��������������Զ���Ů�����������������������90%�����Ի����ĺ���Ϊ46,XX�������46,XY������Ƕ���͡������Ի����IJ����в�������������SRY�����ͻ�����λ��������ԭ��������鲢δ֤ʵ�˼��衣���о���ʾ�����ٵ����ʿ���ϵ�����ٶ�������ѪYȾɫ���SRY����[8]��ȷ���������ʸ�̽���ǻ����鷢������������֯��

2. 45,X/46,XY ���ٷ�����ȫ: ������5����Ⱦɫ��Ϊ45,X/46,XY�������з�����ȫ��غ�������״���٣����е��͵�Turner�ۺ������֣����ٻ��߿������ٷʴ�[9]��ͼ4��������߳����������ϹܵIJ������ϵ�����Ϲ������MIS�����ܲ�ȫ������ӳ����¡�������Щ���߿�����غ����֯�����в����Ϊ�δ�ʱ��غͪ��MIS�����Ե�����������ֳ�����Ի������������Ϲ��˻�������һ�ֿ���������Ⱦɫ��Ƕ��ʱ��غ��ķֻ��յ��ӳ٣��������ܲ����㹻��غͪ��MIS���Ӷ�������������ֳ���ֻ�����������ڡ�